Современные технологии позволяют быстро и качественно секвенировать молекулы ДНК и РНК. А вот для определения аминокислотной последовательности белков аналогичных методов до сих пор не было. Но недавно ученые из США предложили метод массового секвенирования белков, в котором удалось совместить некоторые идеи методов секвенирования ДНК нового поколения и масс-спектрометрии. При помощи реакции Эдмана и флуоресценции ученые смогли отделять по одной аминокислоте от белковых цепей и фиксировать происходящие изменения. Новый метод удалось применить и для исследования посттрансляционных модификаций белковых молекул.
С тех пор, как Фредериком Сенгером был предложен первый метод секвенирования ДНК, прошло уже больше 40 лет. За это время технологии секвенирования шагнули далеко вперед, хотя некоторые базовые идеи продолжают использоваться. Собирательно новые методы называют методами секвенирования нового поколения(next generation sequencing, NGS): они позволяют относительно быстро, массово и дешево выполнять качественный и количественный анализ РНК и ДНК (и сильно выигрывают в сравнении со старыми методами). При этом, что важно, некоторые из этих методов позволяют одновременно анализировать сразу много молекул нуклеиновых кислот.
Суть самого распространенного из таких методов заключается в следующем. Нуклеиновые кислоты режутся в произвольных местах на небольшие одноцепочечные кусочки, которые потом достраиваются специально помеченными нуклеотидами по принципу комплементарности так, что присоединение каждого сопровождается флуоресцентной вспышкой. Эти вспышки фиксируются и переводятся в генетический код. После этого уникальные перекрывающиеся фрагменты кода при помощи компьютерного анализа объединяют в более крупные и восстанавливают исходные последовательности. Таким образом, можно секвенировать геном или транскриптом, но не белки, имеющие другую химическую природу, которая не подразумевает возможность комплементарной достройки: в отличие от нуклеотидов, из которых состоят ДНК и РНК и которые могут образовывать комплементарные пары, белки состоят из аминокислот, у которых нет такой «парности».
Тем не менее, можно получить косвенное представление о имеющихся в клетке белках, секвенируя ДНК/РНК и переводя потом этот код в белковый. Более точный белковый анализ обычно подразумевает использование масс-спектрометрии, позволяющей разделить молекулы по массе, «взвесить» и предсказать их первичную структуру, сравнив молекулярный вес с рассчитанными по геномным данным «табличными значениями». Современные технологии предполагают дополнительную фрагментацию белков протеиназами, которые разрезают их специфически между конкретными аминокислотами, так что на деле анализируется не целый белок, а набор фрагментов, на которые он может быть разобран ферментом. Устойчивый для каждого отдельного белка паттерн разрезов тоже позволяет вытянуть дополнительную информацию о его строении.
Являясь очень распространенным и мощным инструментом, масс-спектрометрия имеет и проблемные стороны. В частности, возникают сложности с новыми и посттрансляционно модифицированными белками, для которых табличные значения отсутствуют (так называемое de novo секвенирование). Более подробно про историю, применение и методы протеомики (науки, которая занимается белками) можно почитать в специальном материале на сайте «Биомолекула».
Эдвард Маркотт (Edward M. Marcotte) и его коллеги из Техасского университета в Остине предложили альтернативу существующим решениям и адаптировали методы NGS для прямого секвенирования белков. Ученые заявляют, что новая технология позволяет обрабатывать миллионы молекул параллельно и идентифицировать в смесях даже штучные белки.
По аналогии с нуклеотидным NGS было решено использовать флуоресцентные метки, чтобы различать элементы аминокислотной последовательности белка, но при этом в качестве причины изменения флуоресценции использовалась деградация, а не синтез цепи. Реакция Эдмана позволяет «откусывать» от N-концабелка по одной аминокислоте и анализировать их потом для восстановления всей последовательности. Этот метод был придуман еще в 50-х годах XX века, в его классическом варианте белки закрепляются на подложке, а отвалившиеся аминокислоты смываются и определяются в хроматографе. Цикл можно повторять, пока не станет известна вся белковая последовательность. Минусы этого метода (в частности, трудо- и времязатратность) не позволяют его использовать в крупномасштабных исследованиях.
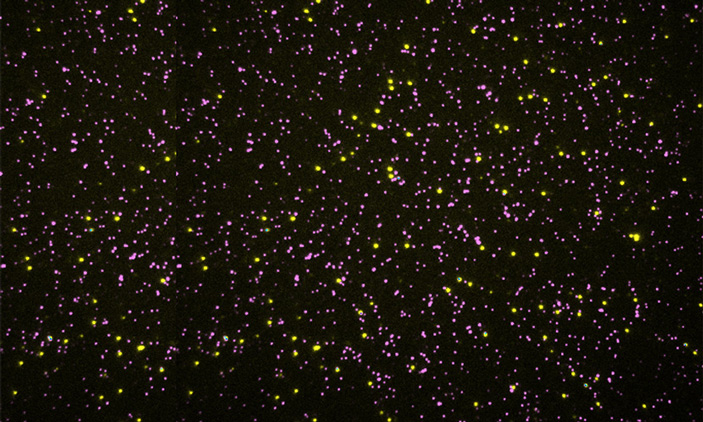
Новая технология отличается от старой тремя важными деталями. Во-первых, на подложке закреплено сразу много разных белков. Это позволяет значительно ускорить процесс, потому что за один цикл удается извлечь информацию об одной хвостовой аминокислоте каждого из них. При этом, если бы ученые продолжили определять последовательность белка классическим способом при помощи хроматографа, они бы зашли в тупик. У разных белков разные концевые аминокислоты, которые перемешивались бы при смытии так, что понять, какая из них принадлежала какому белку, оказалось бы невозможно. Поэтому второе нововведение оказалось связано со способом определения аминокислот — для этого и понадобились флуоресцентные метки. А чтобы избежать их путаницы при смывании, измеряли флуоресценцию не отвалившихся аминокислот, а закрепленных белков. Для этого некоторые аминокислотные остатки белков еще до начала реакции специфически пометили флуорофорами, так, что в камере микроскопа был виден яркий ковер из точек, каждая из которых соответствует закрепленному белку .
Ученые подсчитали, что на шесть с половиной квадратных миллиметров подложки можно впихнуть без потерь для качества три миллиона пептидов. Для сравнения, примерно во столько же оценивается общее число белков и пептидов в одной клетке бактерии кишечной палочки Escherichia coli (по данным из статьи Ron Milo, 2013. What is the total number of protein molecules per cell volume? A call to rethink some published values).
В обсуждаемой работе ученые метили цистеин и иногда лизин или серин, но в принципе возможно подобрать красители и к другим аминокислотам. Если используется метка для одной аминокислоты, все точки получаются одного цвета, а если меток несколько, то цвет зависит от соотношения меток (а значит, и меченых аминокислот) в белке. Стоит заметить, что тут подбираются особенно устойчивые красители — реакция Эдмана использует довольно жесткие реагенты и нагревание, так что нужно чтобы метки не развалились по дороге. Разумеется, они имеют не белковую структуру.
Для эксперимента взяли TIRF-микроскоп с высоким разрешением, позволяющим разглядеть каждую светящуюся точку-пептид. Перед началом реакции уровень и спектр свечения каждой точки замеряется и дальше, по мере протекания реакции съемка микроскопа фиксирует все ее изменения цвета. За один цикл яркость может либо уменьшиться, если последняя аминокислота цепи была меченой и смылась, либо остаться прежней если метки не было. По поэтапному выцветанию точек из одного и того же места определяются положения меченых аминокислот, а по ним вычисляется и сам белок.

Пример результатов, полученных для смеси 1695 молекул пептидов с двумя мечеными цистеинами в разных положениях. Изменение структуры одной из молекул на протяжении восьми циклов реакции (a) и ee фотографии для каждого цикла (b). d-суммарные результаты для всех молекул смеси. Цветом обозначена интенсивность свечения молекул на каждом из восьми циклов реакции. Результаты сгруппированы по сходству и подсчитано количество пептидов, для которых интенсивность свечения упала в одних и тех же циклах, — соответственно, позиции цистеинов в этих молекулах совпадают. Выяснилось, что больше всего в смеси молекул с цистеином во второй и пятой позициях (675 штук). Рисунок из обсуждаемой статьи в Nature Biotechnology
Третья модификация классического метода Эдмана заключалась в том, что белки перед началом реакции режутся на небольшие пептиды, так что в камере закреплены не длинные молекулы, а их кусочки. Сделано это было по нескольким причинам. Во-первых, из-за периодических ошибок в ходе реакции (не сработали реагенты, не смылась аминокислота и т. д.) с каждым циклом накапливается все больше и больше белков, которые находятся на неправильной стадии реакции и, соответственно, неправильно интерпретируются. Разрезание дает возможность проанализировать за небольшое количество циклов весь белок по кусочкам, а не постепенно разбирать всю последовательность. Это избавляет от накапливающихся ошибок и заодно (ведь белки могут быть длиной в несколько тысяч аминокислот) многократно снижает необходимое число циклов реакции.
С другой стороны, такой ход усложният задачу определения: при разрезании информация о целом белке теряется и разрезанные кусочки надо теперь как-то «склеивать» обратно. Исследователи позаимствовали решение этой проблемы у метода масс-спектрометрии. Если разрезы будут происходить не случайным образом (как в случае обычного NGS), а только между конкретными аминокислотами, то у всех пептидов окажутся известные заранее хвосты. Поэтому подбор специфичных протеаз увеличивает количество «символов», которые можно прочитать с белковой молекулы.
Остаточные проблемы, связанные с тем, что белки разрезаны на кусочки и что в них известны не все аминокислоты, решаются за счет грамотного компьютерного анализа результатов. Как и масс-спектрометрия, этот метод пока не может обходиться без заранее созданных баз данных белков, которые могут оказаться в смеси. Найденный участок примеряется на совпадение с подготовленными последовательностями этих белков (чтобы они подходили по формату к экспериментальным данным, для них моделируют места разрезов и положения флуоресцентных меток). Если он подходит, то пробелы между мечеными аминокислотами заполняются из базы данных. Ученые подсчитали, что данных от двух флуоресцентных маркеров и протеиназы должно хватить для разбора смесей средней сложности (около тысячи разных белков), а разработки еще пары меток должно хватить для правильного определения большинства белков протеома.
Исследователи также проверили этот метод для определения посттрансляционных модификаций аминокислот, которые происходят уже после синтеза и никак не отражены в геноме. В качестве наглядного примера они взяли короткую повторяющуюся аминокислотную последовательность, расположенную на хвосте фермента РНК-полимеразы ll (см. CTD of RNA polymerase). Это очень важный для работы полимеразы участок, в котором фосфорилирование разных серинов может провоцировать сплайсинг или кэпирование (J.-P. Hsin, J. L. Manley, 2012. The RNA polymerase II CTD coordinates transcription and RNA processing). Взяв смесь по-разному модифицированных пептидов и пометив фосфосерины, они обнаружили их вымывание на разных циклах реакции Эдмана. Восстановив на основе этого положение фосфорилированных серинов в пептидах, ученые подтвердили потенциальную применимость метода для анализа посттрансляционных модификаций.
Было бы неверно говорить о том, что новый метод решает все проблемы протеомики. Но у него есть важные преимущества перед масс-спектрометрией — высокая чувствительность и параллельное секвенирование многих белков, которые в дальнейшем могут сильно уменьшить трудоемкость этого процесса. Тем не менее, одна из ключевых проблем протеомики — невозможность легкого определения белков de novo — осталась нерешенной. Поскольку метод совсем новый, для него пока не создан промышленный секвенатор, который позволил бы другим исследователям оценить все плюсы и минусы. Но авторы работы планируют дорабатывать метод и создать такой прибора в будущем.
Вера Мухина